Research Overview
Research in the Montagna Laboratory at the Rutgers Cancer Institute aims to understand the mechanisms by which genomic instability promotes tumor initiation and progression. Using advanced molecular cytogenetic approaches and single cell genomics methodologies applied to human and murine in culture and in vivo models we investigate the roles of large copy number alterations and papilloma virus insertional mutagenesis to tumor initiation and progression.
Aneuploidy in aging and cancer risk
Age is the single biggest risk factor for cancer. Genome instability has long been implicated as the main causal factor of aging and it is a hallmark of cancer (Andriani et al.). An active area of investigation in the laboratory is to elucidate the molecular mechanisms by which age-related increased genomic instability leads to increased cancer risk. In previous studies, we uncovered unexpectedly high levels of age-related accumulation of whole chromosome aneuploidy in the aging mouse brain (Faggioli et al., Andriani et al.). We showed that aneuploidy is sufficient to induce senescence, a cell cycle arrest often accompanied by a senescence associated secretory phenotype (SASP) which can alter the surrounding microenvironment (Andriani et al.). Using single cell genomic analysis (Andriani et al., Faggioli et al.) of postmortem human brain tissues our current work aims to define the cells and the brain areas in which genomic instability arise during aging and study how this process increases cancer risk. These experiments combined with in culture 3D cortical spheroids and murine models in which the fate of aneuploid brain cell types can be traced are expected to define the contribution of aneuploidy to disease, establish the molecular pathways altered in the presence of aneuploid cells and investigate if and how somatic cells with an abnormal chromosome number are identified and eliminated (Baker and Montagna).
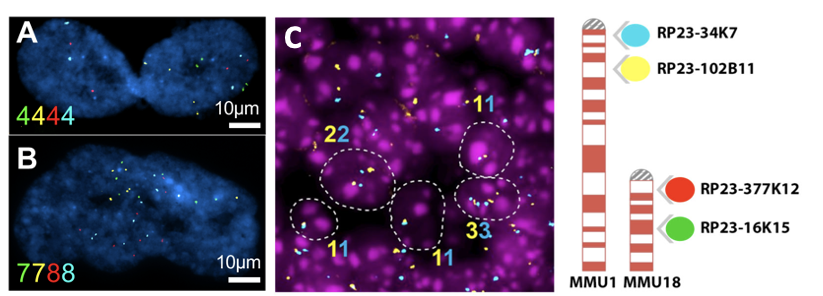
Aneuploidy quantification by Fluorescent in situ Hybridization (FISH). (A-B): Representative images of senescent cells IMR-90 cells (an adherent diploid primary human fibroblast strain from fetal lung). Color numbers indicate the copies for chromosome 9 (green and yellow) and 12 (red and blue). (C) Representative image of FISH in the adult mouse brain sections (left). Ideogram of the 4-color iFISH assay (right).
Haploinsufficiency of BRCA1 and BRCA2 functions and tumor risk
Inherited germline mutations in the BRCA1 or BRCA2 genes cause premature aging phenotypes and greatly increase the risk of breast and ovarian cancer, presumably as consequence of deficient DNA repair, by elevating somatic mutational errors. Using single cell whole genome sequencing technologies we previously established the somatic mutational landscape of primary, non-cancer, mammary epithelial cells of women diagnosed with pathogenic BRCA1 or BRCA2 germline mutations (Sun et al.). Using single cell genomic analyses combined with custom Fluorescent in situ Hybridization (FISH) assays and 3D atmospheres established from mastectomies of germline mutation carriers and controls we aim to define the genomic landscape of mutant cells in response to modifiers of penetrance. Projects are ongoing to define the role of hormonal exposure on mutation rate.
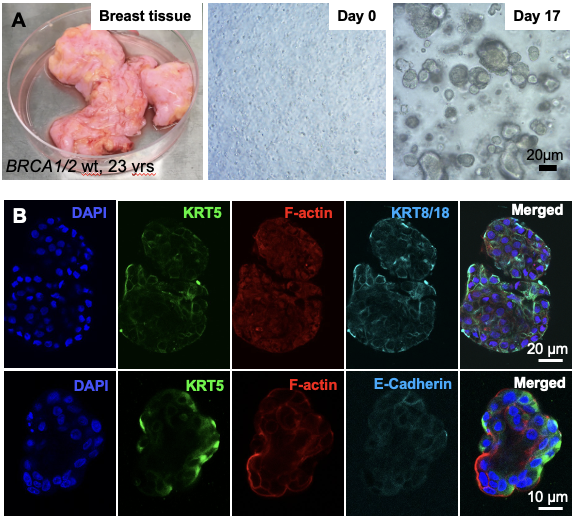
In culture 3D primary human organoids. (A) Process of primary tissue dissociation and establishment of 3D mammospheres. (B) Whole-mount 3D confocal optical sections of a normal human breast organoid derived from admixed basal and luminal progenitor cells labeled for keratin 5 (K5), F-actin, E-Cadherin and 4',6-diamidino-2-phenylindole (DAPI). Scale bar = 20 μm (B) and 10 μm (C).
Papilloma virus insertional mutagenesis to tumorigenesis.
Most cervical cancers are caused by human papillomavirus (HPV). HPV is also the etiological agent that promotes tumorigenesis in a fraction of other types of cancer (e.g., head and neck, vulvar, anal and penile cancer). High-risk HPV types infect epithelial cells of the cervix where the viral DNA genome replicates as an episome. In most cervical tumors and in a subset of other HPV associate cancers, HPV DNA is integrated into human genomic DNA. Although likely infrequent events, HPV DNA integrations can cause stable association of the oncogenic viral E6 and E7 genes with a host cell and its descendents, result in the virus DNA causing alterations in transcription of human cancer genes flanking the insertion sites, and somehow cause rearrangements of human genomic DNA near the inserted HPV DNA, thus contributing to tumorigenesis in multiple ways. The Montagna lab has developed methodologies to detect and map HPV-human DNA junctions at single nucleotide resolution and established molecular cytogenetic approaches to study the clonal evolution of cells with integrated HPV DNA (Van Arsdale et al.). Studies are ongoing to define clonal expansion of cells with integrated HPV in histological tissue samples of premalignant cervical lesions, and to study the functional consequences of HPV DNA integration on cell phenotypes and cell fates.
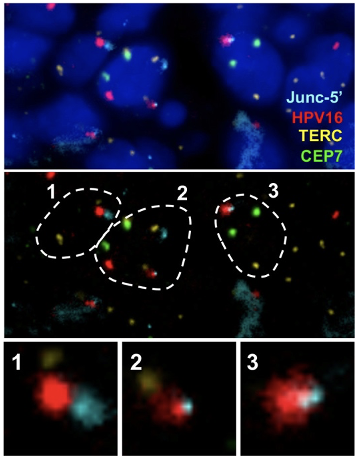 Visualization of HPV16 insertion by Junc-FISH in a CIN3 sample. An HPV16 insertion detected in a CIN3 sample by HC+NGS identified a viral DNA insertion between Chr2:125,862,000 and 125,911,000, accompanied by a ~49kb deletion of human sequences. Custom FISH probes were generated using Bacterial Artificial Chromosome (BAC) RP11-632F4 (Junc-5’) on Chr2:125,741,932-125,918,993 spanning the integration mapped by HC+NGS. Probes for the telomerase RNA component (TERC) gene reported as amplified in cervical cancer as well as a chromosome 7 enumeration probe (CEP7) were also included.
Visualization of HPV16 insertion by Junc-FISH in a CIN3 sample. An HPV16 insertion detected in a CIN3 sample by HC+NGS identified a viral DNA insertion between Chr2:125,862,000 and 125,911,000, accompanied by a ~49kb deletion of human sequences. Custom FISH probes were generated using Bacterial Artificial Chromosome (BAC) RP11-632F4 (Junc-5’) on Chr2:125,741,932-125,918,993 spanning the integration mapped by HC+NGS. Probes for the telomerase RNA component (TERC) gene reported as amplified in cervical cancer as well as a chromosome 7 enumeration probe (CEP7) were also included.
Top: representative nuclei depicting DAPI, Chr2 Junc-5’, HPV16, TERC and CEP7. Middle: outline of the same 3 cells (1-3) where the HPV16 probe colocalized with the Chr2 Junc-5’ probe. TERC and CEP7 are also shown. (C) enlargement of the colocalized signals which include FISH signals specific for HPV16 overlapping with the Chr2 Junc-5’ BAC clone corresponding to the region on Chr2 where the insertion was mapped.
Septin 9 (SEPT9) oncogene amplification promotes invasion
SEPT9 is a member of a family of filamentous GTP-binding proteins that assembles into filaments and rings. We discovered that some breast tumors harbor genomic amplification of Sept9 in the form of acentric chromosomal fragments known as double minutes (dmin) chromosomes, extra-chromosomal bodies of circular DNA containing mainly oncogenes (Montagna et al.). In our previous work we found that SEPT9 assembled into filaments that colocalize with invadopodia and FAs and that overlapped with the gelatin degradation patterns implying that the ECM degradation may be, at least in part, mediated by SEPT9 (Marcus et al.). Work is ongoing to establish the mechanism by which SEPT9 amplification and over expression promotes tumor cell invasion in metastatic carcinomas

SEPT9 and MMP3 expression in mammary epithelial cells. SEPT9-GFP and MMP3 expression in MDA-231 grown on gelatin coated coverslips. MMP3 is visible in vesicles-like structures localize at the invasive front of the depicted MDA-231 cell. MMP3 expression is visible along SEPT9 filaments.
Research in the Montagna laboratory is supported by The National Institutes of Health (NIH) and The Department of Defense (DoD) Breast Cancer and Ovarian Research Program.